{kind=link}
{kind=link}
{kind=link}
{kind=link}
Collaboration between the Galaxy and VGP communities to translate the Sanger TreeVal NF DDL workflow into something equivalent or better in Galaxy. Everyone with an interest in contributing to this effort is cordially invited to pitch in.
Origins and early progress documented back here

These are from the current version that has a couple of smaller subworkflows - TreeValGalSimilarity.ga TreeValGal_anno_bed_optional.ga and TreeValGal_bed_to_bigwig.ga
TreeValGal depends on the updated Jbrowse2 tool to present repeats, gaps and other features as tracks in a configurable browser.
JBrowse2 is currently only available on usegalaxy.eu for testing.
The workflow will soon be added to the VGP workflow repository where development will continue after May 1 2024.

This repository will be left for the historical record if anyone is interested, since it contains an account of implementing the features of a NextFlow workflow as Galaxy workflows. One of the main differences is that Galaxy workflows and subworkflows are built by dropping, configuring and connecting tools on a graphical canvas, without requiring any code to be supplied.
To move the output of one NextFlow module into the input of another in any new Nextflow workflow, the developer must examine each module's DDL code and metadata files, to discover what the input and output file names and formats are. Then they must write dedicated DDL code to create the data channels NextFlow needs to connect those two modules correctly. Similarly, to set the parameter values for each module, the workflow developer must read the code and metadata for the module, noting the names and formats for the parameters, and then provide specific DDL and configuration files with the correct values, to set those module variables at run time. The developer is responsible for getting all these details right because there is no automated interface for NextFlow modules.
In contrast, the Galaxy server's abstract tool interface allows it to automatically discover the files and data types needed for any tool. The user draws a "noodle" to join the output and input of any two tools, on a graphical workflow construction canvas. The server permits two tools to be connected only where the data types match or can be converted automatically, preventing users from misconfiguring tool inputs. Similarly, a Galaxy workflow builder does not need to examine the code for each tool to provide appropriate parameter names and values for a workflow. Galaxy generates the tool input form automatically, from each tool's interface configuration document. The workflow developer fills in each tool form as needed, without any of the internal tool details. Input data files, and run time parameters are added by dropping and configuring workflow input and control tools onto the canvas. Any available Galaxy tool can be dropped onto any workflow canvas. Any data input or individual tool parameter can be turned into a workflow form field for the user to set. All Galaxy tools are interoperable with no additional programming in workflows. Tool parameter and data connection settings are saved in a server generated, shareable JSON document representing the entire workflow. The Galaxy tool abstract interface supports automated tool interoperability, supporting efficient development and improvement of complex workflows without any specialised coding.
Some work on sequence similarity mapping for haplotypes suggests that automated harmonisation of haplotype strand and contig order/naming could be possible early. With an external reference, the "truth" is known, so only the haplotype fasta ordering/naming should be changed, but when the two haplotypes are used, the choice is arbitrary and these could be reordered on one or the other axis to reclaim the latent expected diagonal line of similarity between the contigs.
Could simplify situations where comparable information from both haplotypes could be used to help resolve ambiguities in either - at least for the very largest contigs with strong sequence similarity between the two haplotypes.
- How best to handle variable number of AA/RNA/CDS/gene NCBI annotation files?
- may be no useful one or many
- cannot see how make minimap or miniprot optional in a WF, with an optional input
- tool insists on a non-optional input - doesn't know is running in a WF
- can make them optional as JB2 tracks
- Available ones selected from history
- Must be already mapped as GFF with miniprot for AA and as minimap BAM for the others
- Mashmap is stalling, spinning forever in workflows.
- May need additional resources.
- Self-homology mapping tracks prove problematic
- Sanger use mummer - a suffix tree mapper. source untouched for >4 years.
- max 500k contigs - much flailing to reassemble after cutting and mapping - yumi sims code.
- minimap has a special mode
- generates many hits and would need filtering
- Should low complexity regions be masked?
- ignore likely non-informative "homology" in repetitive sequence?
- Anna working on nucleotide and peptide alignments subworkflows
- ncbi downloader improvements suggested
- Next JB2 release has optional inputs for workflows.
- New AutogenJB2 (collection -> JB2) tool soon available on EU
- The core of the JB2 track generator requires only a path, name and ext in most cases
- paf will need a subcollection of paf, reference1...referenceN
- The core of the JB2 track generator requires only a path, name and ext in most cases
-
Long term goal
- Add interactive JBrowse configurations to genomeArk web pages, to make VGP data easily accessible as tracks to anyone with a web browser.
-
Intermediate milestones
- Production TreeValGal for internal VGP use
- Build and distribute other VGP JB2 track sets for different audiences
- Provide a test bed for the development and display of automated annotation
- integrate useful tracks into TreeValGal and other outputs as they become available.
- Additional JBrowse2 GTN training modules
- create dot plots and reverse mappings from mashmap PAF tracks ...
-
Short term milestones
- a "good to test" track list with tool tuning parameters as needed.
- Implement as the initial TreeValGal workflow and hand over
- Clean-up and acceptance-testing as part of a routine VGP WF run.
- Busco genes could be added to TreeValGal as GFF3 if available
- Action: could someone please make a sample busco gff3 for one of the test assemblies we are currently using: calanna and bralanc ?
- a gff is probably more suited for a JB2 track than a bed
- easy enough to include both if useful
-
The next release (_5) of JBrowse2 on EU will optionally create a zip file instead of being a viewable HTML page.
- This enables the Galaxy uploader to push a JBrowse directory to S3. It seemed confused by html
- Public website proof of concept using a zip from Galaxy to an S3 bucket thanks to Bjoern.
- This enables the Galaxy uploader to push a JBrowse directory to S3. It seemed confused by html
-
Nadolina has nominated the gene_alignment TreeVal subworkflow to implement.
- Tools should all be on EU but may have missed something.
- Two main subworkflows are used - for nucleotide and peptide mapping so each of those can be built and tested
- Once tested, we can figure out how best to fit in with the existing WF.
-
mashmap proposed as a way to make multi-organism PAF tracks.
- Ross built and tested a prototype
- Bjoern has take the tool over, and made it far better.
- Will soon be available from IUC
- Sample 3 way PAF display below was manually set up, starting with a JBrowse with a single mashmap track and arabidopsis reference.
- Other two genomes added as separate linear tracks below the main reference gives reverse mappings!
- Easy to configure with JB2 - a few moments clicking - all the references are available.
- Dot plots too - all there but require manual exposure

Here are some bird on bird dot plots - they even look a bit like a flock of birds if you squint.

- What additional tracks would be useful?
- for VGP internal use?
- Other TreeVal modules: selfcomp
- kmer/busco/pretext etc already in VGP)
- Other TreeVal modules: selfcomp
- for external researchers
- for the public - making VGP data really easy to "access"
- for VGP internal use?
- Track files being generated routinely can be added if there's a URI or path.
- Which of the existing VGP WF outputs would be useful to add?
- tracks take some disk space but there's no real technical limit to the size of a track menu.
- Which of the existing VGP WF outputs would be useful to add?
- Other VGP assemblies to run TreeValGal on to serve as samples?
- JBrowse2 directories support interactive display
- Any byte-range request static web server like nginx or apache.
- Can also view on a local laptop browser without Galaxy or internet access
- a tiny pop-up python webserver
jb2_webserver.pyis included with the data for local displays.
- a tiny pop-up python webserver
- How to allow access to big JBrowse2 configurations efficiently?
- JBrowse2 archives contain compressed/indexed reference sequence and track files - so are big.
Anna asked about a repeatmasker version of the repeats, so there's a gff track (no dfam species and defaults) to compare to the windowmasker bed and wiggle on the current Amphioxus JBrowse2.

Repeatmasker seems more selective with half or fewer but bigger hits. It is model based so takes much longer (~2 orders of magnitude) to complete although that's not important for a workflow. 7 hours 4 cores vs 15 minutes 1 core for the hummingbird for example.
The other repeat data is from a model-free method - NCBI windowmasker - so no surprise that a model-based method like repeatmasker is verrrrry different with defaults - suggestions appreciated!
Here's a zoomed in part of the current hummingbird TreeValGal output after hiding the other tracks and opening the repeatmasker gff and the repeat bed. The wiggle is windowmaker smoothed over 1k windows and reflects that detail differently, particularly when zoomed in.

Each method and view gives different information about the complex underlying biology here and JBrowse2 makes it easy to pick and choose between them.

Generating and publishing updated live JBrowse2 links for any VGP page is possible, because JBrowse2 configuration can be completely machine driven.
- Offer convenient, highly granular tracks derived from VGP and other data
- readily accessible to anybody with internet, a web browser and a question.
- Adding automated feature annotation tracks could build a potentially valuable, accessible and sustainable resource for genome sciences.
Each JBrowse2 directory is independent, with an index.html, track files and javascript.
- served typically by Apache or Nginx
- potentially stored (untested!) on an S3 bucket.
Most of the components are available.
- An initial functional track menu can quickly be made available.
- It will require storage and effort TBA
- Proof of concept sample outputs below.
Challenges.
- storage for compressed/indexed data (a few GB each perhaps)
- computational resources required are very substantial for creation.
- Individual updates are only needed when data changes.
- Workflow change means recreating everything
- not a problem if "reuse jobs" really works...
- ongoing dedicated effort for sustainable development.
Given those, configurations can be improved, by editing the workflows, to suit real user needs. The rat database does this with JB1, so it's not novel, but would add a lot of value to existing data with relatively little additional resources.

Now does 10 track types - any of these datatypes in a Galaxy history or workflow can be added as a track to a JBrowse2 instance. Outputs are immediately viewable in Galaxy. The JBrowse2 directory can be downloaded as a zip file, for local unzipping and viewing using a provided tiny python web server. These can also be served using any byte-range request static web server. They tend to be huge - GB in size, with the reference and all tracks in compressed and indexed formats.
bam
bed
bigwig
blastxml to gff3
cram
gff3
hic
maf
paf
vcf
| Module | Status |
|---|---|
| yaml_input | Not needed |
| gap_finder | Prototype available |
| generate_genome | Not needed Existing chromosome lengths tool works in one step. |
| hic_mapping | Fixing hicBuildMatrix for a cool matrix from the VGP hic workflows |
| kmer | Awaiting fastk and merquryfk tool wrappers |
| longread_coverage | Partial prototype available |
| nuc_alignments | |
| pep_alignments | |
| punchlist | Need help - part of hic generation |
| repeat_density | Prototype available. |
| synteny | Prototype available. |
| telo_finder | Prototype available in treevalgal workflow now using seqtk-telo |
Updating JBrowse1 tool code to JBrowse2 over the past 5 weeks. These are 8 samples - paf and hic are also available.

Working well, but not in IUC yet. Alternative PR is being resurrected so may take some time.
Deployed on EU as a test tool. See treevalgal workflow for more details. Broken wiggle and paf labels in the samples have been fixed.

- Need fastk/merquryFK for a KMER subworkflow.
- A Fastk wrapper is currently being developed.
- Bjoern is thinking about how best to integrate binary HiC format data into the existing infrastructure.
- Jb2 has specialised Multiple Alignment Format (MAF) tracks, and a blastXML track (translated into GFF3), and those will all be useful in VGP work.
- awaiting a PR merge and update to BCBIO-GFF conda dependency from Brad Chapman to get these activated.
Galaxy's integrated support for genomic feature visualisation at scale will be very hard to match.
With thanks to Bjoern Gruening and Anna Syme for helping with testing and tools, and the support of Galaxy Australia, there is now a WIP TreeValGal workflow on usegalaxy.eu that combines the two gap tracks, the repeats track, a telomere track and the coverage track for the TreeVal small sample test data into a single JBrowse viewer.
Please try it on your own pacbio/refseq data and let me know if this is worth more work to add additional TreeVal tracks to for your use?
Sample images below show how JBrowse does all the work for us. All tracks are also in the history as bed files if the user wants them for downstream analyses.



- Some sample outputs from a prototype longread_coverage workflow under development and testing.
- Some sample outputs from a prototype repeat_density workflow, now available for testing. Not yet working on the usegalaxy servers. Needs the window_masker tool installed.
- See sample outputs from a prototype gap_finder workflow, now available for testing.
- seqtk-telo is now available in the seqtk suite from the Toolshed.
Plans are documented for discussion and refinement at the work plan discussion
For the “rapid” workflow, not all the 18 subworkflows are needed, so this makes a good first target for implementation. The NF DDL shows the main submodules needed:
//
// IMPORT: SUBWORKFLOWS CALLED BY THE MAIN
//
include { YAML_INPUT } from '../subworkflows/local/yaml_input'
include { GENERATE_GENOME } from '../subworkflows/local/generate_genome'
include { REPEAT_DENSITY } from '../subworkflows/local/repeat_density'
include { GAP_FINDER } from '../subworkflows/local/gap_finder'
include { LONGREAD_COVERAGE } from '../subworkflows/local/longread_coverage'
include { TELO_FINDER } from '../subworkflows/local/telo_finder'
include { HIC_MAPPING } from '../subworkflows/local/hic_mapping'
include { KMER } from '../subworkflows/local/kmer'
HIC_MAPPING needs some additional subworkflows so the working list for implementation is as above
A first pass at documenting each subworkflow at the level of dependencies and command lines executed at each step has been completed ready for comment and review. While clarifying the best long term solutions, prototype TreeVal Galaxy subworkflows can be created with existing and some new tools, so users can test them. These prototypes will help drive the design and features of an optimal production workflow, as discussions progress.
Anton has nominated TreeVal as a test case for translating a NextFlow (NF) workflow into Galaxy.
That translation process needs a clear understanding of the parameters and data TreeVal needs from the user, and what specific application, data and command line is executed at each step.
Once understood, workflows that re-create the same logic can be built using existing or new Galaxy tools, combined into Galaxy subworkflows and a complete Galaxy workflow implemented, tested and documented.
In the longer term, this is intended to serve as a prototype, to allow users to test it on their own data. Reaching consensus on an optimal design when starting from scratch, may be a challenging path, but once all the needed tools are available, Galaxy's GUI workflow editing makes it easy to implement ideas from users trying things out to incrementally improve working workflows.
Expertise and effort will be needed for:
- NF DDL for insight into how the workflow works
- Warning! The DDL analysis here is guesswork by Galaxy collaborators, who have never actually used NF!
- It needs to be reviewed properly.
- the specific scientific context for checking data flows and testing new tools,
- tool and workflow technical skills for building, testing and documentation.
Sanger publish usage and technical documentation for the TreeVal workflow source code. There is an overview flow diagram, detailed output description for each subworkflow, and an online NF setup generator.
{kind=link}
Test data is available using:
curl https://tolit.cog.sanger.ac.uk/test-data/resources/treeval/TreeValTinyData.tar.gz | tar xzf -
The tiny test data is 1.2GB
Understanding the NF DDL used to write workflows and modules is key to replicating the functionality needed for Galaxy. Content expertise is also a key ingredient because it can help understand how to adapt NF idioms and idiosyncrasies for Galaxy, confirm process logic, and to test and validate tools and subworkflows as they become available.
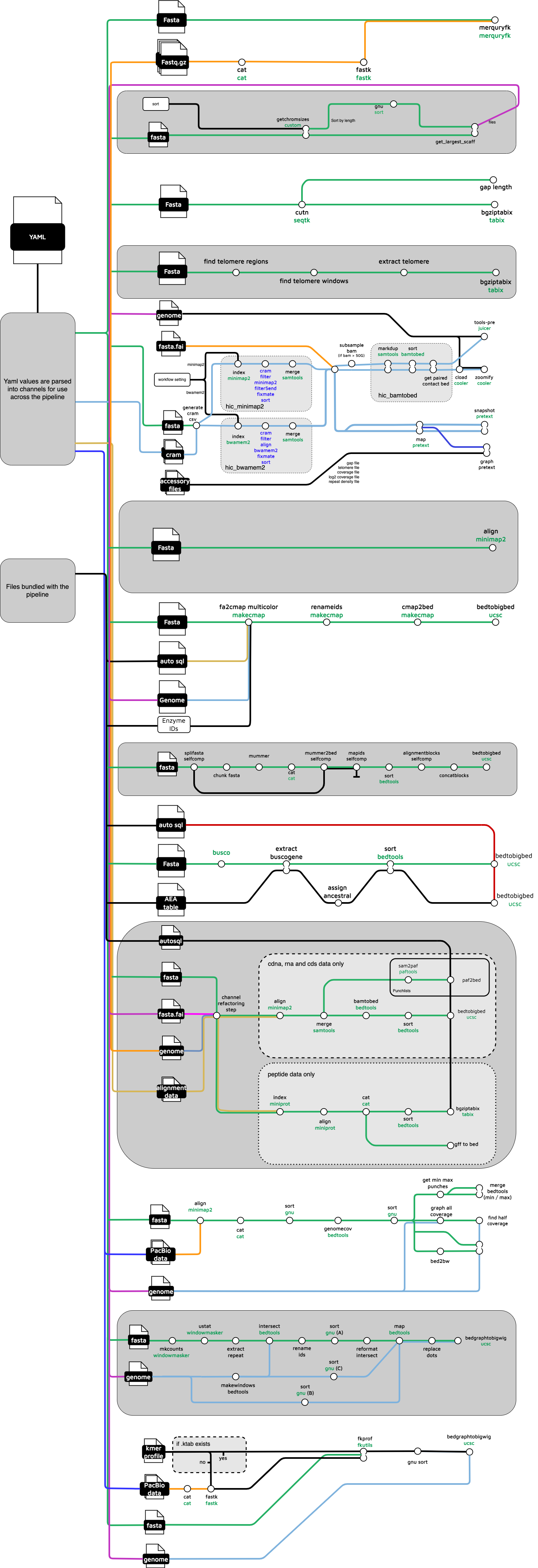
NF workflows contain calls to subworkflows and modules. The functionality of a single NF module is usually the Galaxy equivalent of a tool. There are 2 TreeVal workflows . The full one has 18 subworkflows. They depend on 20 NF-core modules and 35 local workflow-specific NF DDL modules that depend on 22 specialised scripts and jar files supplied in the /tree/bin directory. The “rapid” workflow uses only a subset of those subworkflows.
Parameter preparation for module command lines is integrated into the NF workflow DDL. In particular, subworkflows often seem to have specialised DDL involving data and parameters as “channels”, to allow modules to be chained, in contrast to tools connected with noodles in the Galaxy workflow construction GUI.
Galaxy tools are configured to control third-party analysis application code by a wrapper XML document. Applications are hidden from the server behind an abstract tool interface. This isolation layer allows the Galaxy workflow user to control data and parameter requirements for any tool, using familiar input widgets and explanatory text, without additional programming. Data flow between any two tools is controlled by connecting an input to an output with matching datatypes in a GUI - no additional programming is required.
As a first try, it makes sense to more or less re-use the NF subworkflow and module logic in Galaxy. Without content expert guidance, there is no information to guide improvements to the existing workflow logic, so the goal is to copy it and duplicate functionality. Optimisation for Galaxy users then becomes possible because users can respond to the working workflow. It may be possible to build complicated Galaxy tools to perform many steps in a subworkflow, but individual components may be re-used in other parts of related pipelines, so one tool per module may be the most adaptable initial approach.
Note that every new tool written will require effort for maintenance. Some will be needed but it is recommended that where it is possible, the preference will always be to choose an existing, known-good tool for the new subworkflows if it is possible with minor tweaks in the subworkflow itself.
Note: TreeVal uses tabix at the end of many subworkflows. This may be so the outputs can be viewed using Jbrowse. Galaxy's Jbrowse does the compression and indexing automatically so there is no need for that step unless the tabix file is used downstream for some other subworkflow.
The first target is the rapid workflow described above. Once that is done, new Galaxy subworkflows preserving the logic in the [full NF Sanger workflow](https://raw.githubusercontent.com/sanger-tol/treeval/dev/docs/images/v1-1-0/treeval_1_1_0_full_diagram.png), some **_50+ components will be needed_**. Each more or less corresponds to a Galaxy tool, so the workflow will probably involve 50+ individual tools.For the full workflow, the subworkflows to be translated into Galaxy are listed below. As each one is documented, a link will indicate that there is useful material available for that subworkflow.
- yaml_input
- ancestral_gene
- busco_annotation
- gap_finder
- gene_alignment
- generate_genome
- hic_mapping
- insilico_digest
- kmer
- longread_coverage
- nuc_alignments
- pep_alignments
- pretext_ingestion
- punchlist
- repeat_density
- selfcomp
- synteny
- telo_finder