03. DNA mapping
The snakemake pipeline requires at least two files: a) the .snakefile, containing all the rules that will be run; b) the configuration.yaml file, in which the user can define and customize all the parameters for the different pipeline steps.
Hereafter, the running commands for DNA-mapping and ChIP-seq peak calling will be described for both single and paired-end data.
This short pipeline performs the mapping into a reference genome upon trimming of the raw fastq reads by cutadapt. Further, a filter on the mapping quality (MAPQ) is applied and duplicated reads are marked. Notice that in the case of paired-end reads, when present, UMIs (Unique Molecule Identifiers) sequence is added to the indexes ones in the read name. This is allows the marking of the duplicated reads in a UMI-aware manner (reads/fragments that have exactly the same sequence but different UMI-sequence are not marked as duplicates).
Few information must be provided to the pipeline:
- the source fastq directory
- the output directory where you want your results to be stored (if not already available, the pipeline will make it for you)
- whether your data are paired- or single-end
- the genome to use
All the other parameters are already available in the configfile_DNAmapping.yaml.
| Parameter | Description |
|---|---|
| umi_present | Default: True. True/False to indicate whether the data contain UMIs (ignored for single-end data). |
| fastq_suffix | Default: ".fastq.gz". String with the suffix of the source fastq files. |
| read_suffix | Default: ['_R1', '_R2']. A python-formatted list with two strings containing the suffix used to indicate read1 and read2 respectively. In the case of single end reads, only the first value will be read. If your single data do not have any read-prefix set this parameter to: ['', ''] (blank). |
| cutadapt_trimm_options | Default: '' (blank). String indicating additional user-specific values to pass to cutadapt. |
| fw_adapter_sequence | Default: "AGATCGGAAGAGC". Sequence of the adapter1 (flag -a of cutadapt). |
| rv_adapter_sequence | Default: "AGATCGGAAGAGC". Sequence of the adapter2 (flag -A of cutadapt). |
| run_fastq_qc | Default: False. True/False to indicate whether to run the fastQC on the . |
| use_bwamem2 | Default: False. True/False to define whether to run bwa-mem2 instead of bwa. |
| bwa_options | Default: '' (blank). String indicating additional user-specific values to pass to bwa. |
| remove_duplicates | Default: False. True/False to define whether remove the duplicates from the bam files (if true the tag in the bams will be _dedup instead of _mdup). |
| MAPQ_threshold | Default: 20. All reads with a mapping quality (MAPQ) score lower than this value will be filtered out from the bam files. |
To partially avoid unexpected errors during the execution of the pipeline, a so called 'dry-run' is strongly recommended. Indeed, adding a -n flag at the end of the snakemake running command will allow snakemake to check that all links and file/parameters dependencies are satisfied before to run the "real" processes. This command will therefore help the debugging process.
Always activate your environment, otherwise the pipeline won't be able to find the packages required for the analyses.
snakemake \
--cores 20 \
-s </target/folder>/SPACCa/workflow/SPACCa_DNAmapping.snakefile \
--configfile </target/folder>/SPACCa/config/SPACCa_DNAmapping_configfile.yaml \
--config \
fastq_directory="/path/to/pairedEnd/fastq_data" \
output_directory="/path/to/results/directory/" \
paired_end="True" \
umi_present="True" \
genome_fasta="path/to/genome.fa" \
-nIf no errors occur, the pipeline can be run with the same command but without the final -n flag:
Notice that the absence of errors does not mean that the pipeline will run without any issues; the "dry-run" is only checking whether all the resources are available.
snakemake \
--cores 20 \
-s </target/folder>/SPACCa/workflow/SPACCa_DNAmapping.snakefile \
--configfile </target/folder>/SPACCa/config/SPACCa_DNAmapping_configfile.yaml \
--config \
fastq_directory="/path/to/singleEnd/fastq_data" \
output_directory="/path/to/results/directory/" \
paired_end="False" \
umi_present="False" \
genome_fasta="path/to/genome.fa" \
-nIf no errors occur, the pipeline can be run with the same command but without the final -n flag:
Notice that the absence of errors does not mean that the pipeline will run without any issues; the "dry-run" is only checking whether all the resources are available.
Here after you can see the full potential workflow of the single-end and paired-end DNA-mapping pipeline:
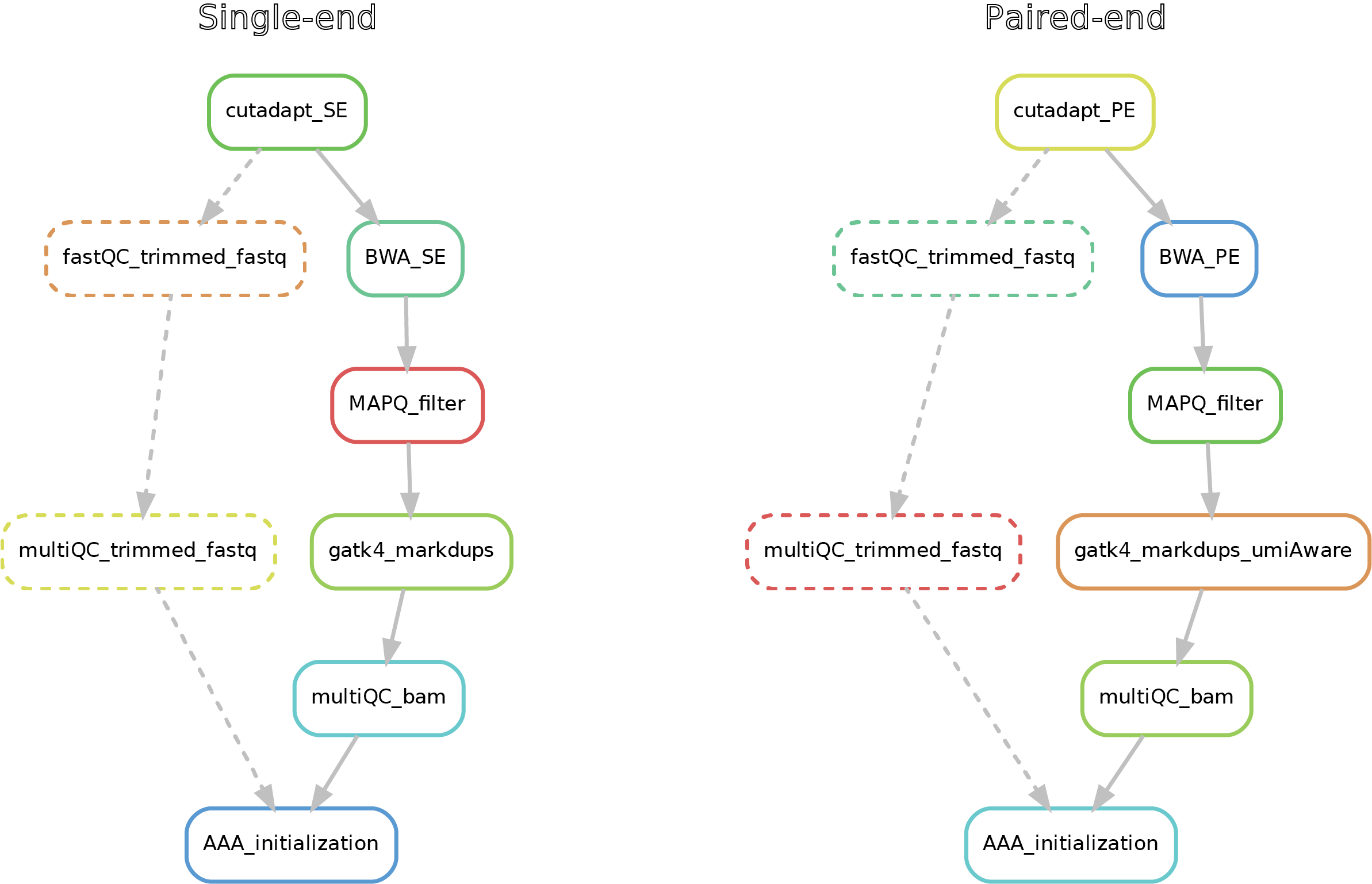
The results structure is the following:
- 01_trimmed_fastq -> fastq.gz files that underwent trimming by cutadapt
- 02_BAM -> mapped reads (bam) filtered for MAPQ, mate-fixed, duplicates marked and eventually UMI-fixed
- 03_quality_controls -> here you can find the fastQC on the trimmed fastq (if required), with the corresponding multiQC report, as well the multiQC report (flagstat + MarkDuplicates) for the filtered bams
Here an example directory tree (paired-end run):
output_folder
├── 01_trimmed_fastq
│ ├── sample_R1_trimmed.fastq.gz
│ └── sample_R2_trimmed.fastq.gz
│
├── 02_BAM
│ ├── sample_mapq20_mdup_sorted.bam
│ ├── sample_mapq20_mdup_sorted.bai
│ ├── BWA_summary
│ │ └── sample.BWA_summary.txt
│ ├── flagstat
│ │ └── sample_mapq20_mdup_sorted_flagstat.txt
| ├── MarkDuplicates_metrics
│ │ └── sample_MarkDuplicates_metrics.txt
│ └── umi_metrics ### (if UMI present) ##
│ └── sample_UMI_metrics.txt
|
└── 03_quality_controls
├── multiQC_bam_filtered
│ └── multiQC_bam_filtered.html
├── trimmed_fastq_fastqc
│ ├── sample_R1_trimmed_fastqc.html
│ ├── sample_R1_trimmed_fastqc.zip
│ ├── sample_R2_trimmed_fastqc.html
│ └── sample_R2_trimmed_fastqc.zip
└── 0trimmed_fastq_multiQC
└── multiQC_report_trimmed_fastq.html
Contributors