A Nextflow pipeline for Variant Calling Analysis with NGS RNA-Seq data based on GATK best practices.

Install Nextflow by using the following command:
curl -s https://get.nextflow.io | bash
Download the Docker image with this command (optional) :
docker pull cbcrg/callings-nf@sha256:b65a7d721b9dd2da07d6bdd7f868b04039860f14fa514add975c59e68614c310
Note: the Docker image contains all the required dependencies except GATK which cannot be included due to license restrictions.
Download the GenomeAnalysisTK.jar (version 3.7) package from this link.
Launch the pipeline execution with the following command:
nextflow run CRG-CNAG/CalliNGS-NF --gatk </path/to/GenomeAnalysisTK.jar>
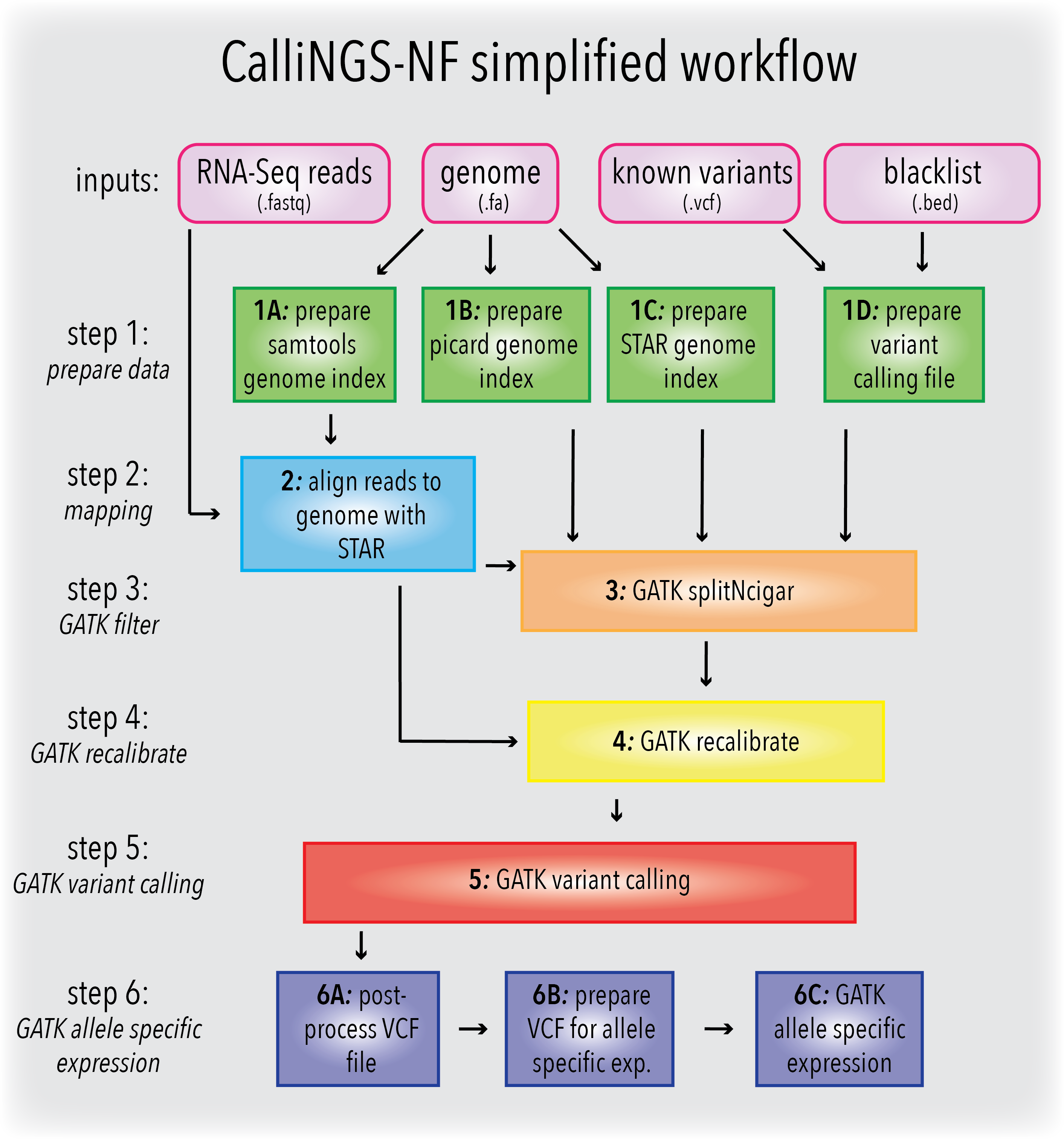
The RNA sequencing (RNA-seq) data, in additional to the expression information, can be used to obtain somatic variants present in the genes of the analysed organism. The CalliNGS-NF pipeline processes RNAseq data to obtain small variants(SNVs), single polymorphisms (SNPs) and small INDELs (insertions, deletions). The pipeline is an implementation of the GATK best practices for variant calling on RNAseq and includes all major steps of the analysis, link.
In addition to the GATK best practics, the pipeline includes steps to compare obtained SNVs with known variants and to calculate allele specific counts for the overlapped SNVs.
The CalliNGS-NF pipeline needs as the input following files:
- RNAseq reads,
*.fastq - Genome assembly,
*.fa - Known variants,
*.vcf - Blacklisted regions of the genome,
*.bed
The RNAseq read file names should match the following naming convention: sampleID{1,2}_{1,2}.extension
where:
- sampleID is the identifier of the sample;
- the first number 1 or 2 is the replicate ID;
- the second number 1 or 2 is the read pair in the paired-end samples;
- extension is the read file name extension eg.
fq,fq.gz,fastq.gz, etc.
example: ENCSR000COQ1_2.fastq.gz.
- Specifies the location of the reads FASTQ file(s).
- Multiple files can be specified using the usual wildcards (*, ?), in this case make sure to surround the parameter string value by single quote characters (see the example below)
- By default it is set to the CalliNGS-NF's location:
$baseDir/data/reads/rep1_{1,2}.fq.gz - See above for naming convention of samples, replicates and pairs read files.
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --reads '/home/dataset/*_{1,2}.fq.gz'
- The location of the genome fasta file.
- It should end in
.fa. - By default it is set to the CalliNGS-NF's location:
$baseDir/data/genome.fa.
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --genome /home/user/my_genome/human.fa
- The location of the known variants VCF file.
- It should end in
.vcforvcf.gz. - By default it is set to the CalliNGS-NF's location:
$baseDir/data/known_variants.vcf.gz.
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --variants /home/user/data/variants.vcf
- The location of the blacklisted genome regions in bed format.
- It should end in
.bed. - By default it is set to the CalliNGS-NF's location:
$baseDir/data/blacklist.bed.
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --blacklist /home/user/data/blacklisted_regions.bed
- Specifies the folder where the results will be stored for the user.
- It does not matter if the folder does not exist.
- By default is set to CalliNGS-NF's folder:
results
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --results /home/user/my_results
- Specifies the location of the GATK jar file.
- Download the
GenomeAnalysisTK.jarpackage from this link. - By default is set to CalliNGS-NF's locations:
/opt/broad/GenomeAnalysisTK.jar.
Example:
$ nextflow run CRG-CNAG/CalliNGS-NF --gatk /opt/broad/GenomeAnalysisTK.jar
For each sample the pipeline creates a folder named sampleID inside the directory specified by using the --results command line option (default: results).
Here is a brief description of output files created for each sample:
| file | description |
|---|---|
final.vcf |
somatic SNVs called from the RNAseq data |
diff.sites_in_files |
comparison of the SNVs from RNAseq data with the set of known variants |
known_snps.vcf |
SNVs that are common between RNAseq calls and known variants |
ASE.tsv |
allele counts at a positions of SNVs (only for common SNVs) |
AF.histogram.pdf |
a histogram plot for allele frequency (only for common SNVs) |
- Nextflow 0.24.x (or higher)
- Java 7/8
- Docker 1.10 (or higher) or Singularity engine
- GATK 3.7
Note: CalliNGS-NF can be used without a container engine by installing in your system all the required software components reported in the following section. See the included Dockerfile for the configuration details.
CalliNGS-NF uses the following software components and tools:
- Java 8
- Picard 2.9.0
- Samtools 1.3.1
- Vcftools 0.1.14
- STAR 2.5.2b
- GATK 3.7
- R 3.1.1
- Awk
- Perl
- Grep