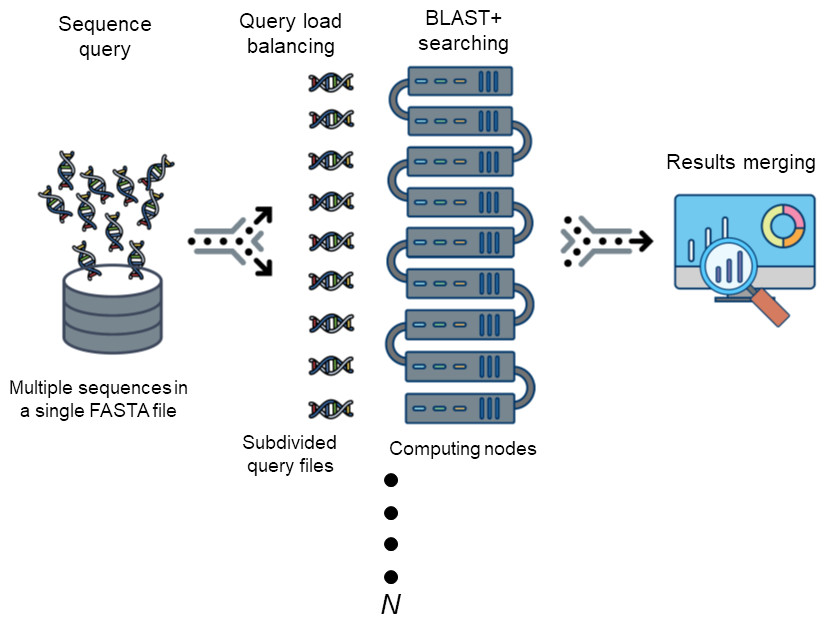
The Basic Local Alignment Search Tool (BLAST) is by far best the most widely used tool in for sequence analysis for rapid sequence similarity searching among nucleic acid or amino acid sequences. Recently, cluster, HPC, grid, and cloud environmentshave been are increasing more widely used and more accessible as high-performance computing systems. Divide and Conquer BLAST (DCBLAST) has been designed to perform run on grid system with query splicing which can run National Center for Biotechnology Information (NCBI) BLAST. BLAST search comparisons over withinthe HPC, grid, and cloud computing grid environment by using a query sequence distribution approach NCBI BLAST. This is a promising tool to accelerate BLAST job dramatically accelerates the execution of BLAST query searches using a simple, accessible, robust, and practical approach.
- DCBLAST can run BLAST job across HPC (SGE & SLURM).
- DCBLAST suppport all NCBI-BLAST+ suite.
- DCBLAST generate exact same NCBI-BLAST+ result.
- DCBLAST can use all options in NCBI-BLAST+ suite.
Following basic softwares are needed to run
- Perl (Any version 5+)
$ which perl
$ perl --version
- NCBI-BLAST+ (Any version; tested 2.4.0+ - 2.7.1+) for easy approach, you can download binary version of blast from below link. ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST
For using recent version, please update BLAST path in config.ini
$ which blastn
- Sun Grid Engine (Any version)
- SLURM (tested with 17.02.2)
- SGE
$ which qsub
- SLURM
$ which sbatch
- Grid cloud or distributed computing system.
The following Perl modules are required:
- Path::Tiny
- Data::Dumper
- Config::Tiny
Install prerequisites with the following command:
$ cpan `cat requirement`
or
$ cpanm `cat requirement`
or
$ cpanm Path::Tiny Data::Dumper Config::Tiny
We strongly recommend to use Perlbrew http://perlbrew.pl/ to avoid having to type sudo
We also recommend to use 'cpanm' https://github.com/miyagawa/cpanminus
The program is a single file Perl scripts. Copy it into executive directories.
We recommend to copy it on scratch disk.
cd ~/scratch/ # We recommend to copy it on scratch disk.
git clone git://github.com/ascendo/DCBLAST.git
cd ~/scratch/DCBLAST_{your_scheduler_SGE_or_SLURM}
perl dcblast.pl
Usage : dcblast.pl --ini config.ini --input input-fasta --size size-of-group --output output-filename-prefix --blast blast-program-name
--ini <ini filename> ##config file ex)config.ini
--input <input filename> ##query fasta file
--size <output size> ## size of chunks usually all core x 2, if you have 160 core all nodes, you can use 320. please check it to your admin.
--output <output filename> ##output folder name
--blast <blast name> ##blastp, blastx, blastn and etcs.
--dryrun Option will only split fasta file into chunks
[dcblast]
##Name of job (will use for SGE job submission name)
job_name_prefix=dcblast
[blast]
##BLAST options
##BLAST path (your blast+ path); $ which blastn; then remove "blastn"
path=~/bin/blast/ncbi-blast-2.2.30+/bin/
##DB path (build your own BLAST DB)
##example
##makeblastdb -in example/test_db.fas -dbtype nucl (for nucleotide sequence)
##makeblastdb -in example/your-protein-db.fas -dbtype prot (for protein sequence)
db=example/test_db.fas
##Evalue cut-off (See BLAST manual)
evalue=1e-05
##number of threads in each job. If your CPU is AMD it needs to be set 1.
num_threads=2
##Max target sequence output (See BLAST manual)
max_target_seqs=1
##Output format (See BLAST manual)
outfmt=6
##any other option can be add it this area
#matrix=BLOSUM62
#gapopen=11
#gapextend=1
[sge]
##Grid job submission commands
##please check your job submission scripts
##Especially Queue name and Threads option will be different depends on your system
pe=SharedMem 1
M=your@email
o=log
q=common.q
j=yes
cwd=
If you need any other options for your enviroment please contant us or admin
PBS & LSF need simple code hack. If you need it please request through issue.
This sequences are randomly selected from plant species. The size and gene number informations are below.
test_db.fas
Number of gene 35386
Total size of gene 43546761
Longest gene 16182
Shortest gene 22
test_query.fas
Number of gene 6282
Total size of gene 7247997
Longest gene 11577
Shortest gene 22
It usually finish within ~20min depends on HPC status and CPU speed.
perl dcblast.pl
Usage : dcblast.pl --ini config.ini --input input-fasta --size size-of-group --output output-filename-prefix --blast blast-program-name
--ini <ini filename> ##config file ex)config.ini
--input <input filename> ##query fasta file
--size <output size> ## size of chunks usually all core x 2, if you have 160 core in nodes, you can use 320. please check it to your admin.
--output <output filename> ##output folder name
--blast <blast name> ##blastp, blastx, blastn and etcs.
--dryrun Option will only split fasta file into chunks
perl dcblast.pl --ini config.ini --input example/test_query.fas --output test --size 20 --blast blastn --dryrun
DRYRUN COMMAND : [qsub -M your@email -cwd -j yes -o log -pe SharedMem 1 -q common.q -N dcblast_split -t 1-20 dcblast_blastcmd.sh]
DRYRUN COMMAND : [qsub -M your@email -cwd -j yes -o log -pe SharedMem 1 -q common.q -hold_jid dcblast_split -N dcblast_merge dcblast_merge.sh test/results 20]
DRYRUN COMMAND : [qstat]
DONE
Check the test folder "test/chunks/" for sequence split result.
You don't need to run "dryrun" everytime.
perl dcblast.pl --ini config.ini --input example/test_query.fas --output test --size 20 --blast blastn
This run will splits file into 20 chunks, run on 20 cores and generated BLAST output file "test/results/merged" and chunked input file "test/chunks/"
It will finish their search within ~20min depends on HPC status and CPU speed.
For your research, please format database according to NCBI-BLAST+ instruction. Here is the brief examples.
makeblastdb -in your-nucleotide-db.fa -dbtype nucl ###for nucleotide sequence
makeblastdb -in your-protein-db.fas -dbtype prot ###for protein sequence
The authors would like to thank DOE for providing support for this study and acknowledge the support of Research & Innovation and the Office of Information Technology at the University of Nevada, Reno for computing time on the Pronghorn High-Performance Computing Cluster.
Won Cheol Yim and John C. Cushman (2017) Divide and Conquer BLAST: using grid engines to accelerate BLAST and other sequence analysis tools. PeerJ 10.7717/peerj.3486 https://peerj.com/articles/3486/